哈工大报








最新发布
- 03-20
- 03-20
- 03-20
- 03-19
- 03-19
- 03-19
- 03-18
- 03-17
哈工大王亚东教授团队提出多组学通用的细胞丰度推断方法
-
分享
微信扫一扫:分享
哈工大全媒体(阚思邈 赵天意/文 赵天意/图)我校计算学部王亚东教授团队在生物信息学算法研究方面取得重要进展,提出多组学通用的细胞丰度推断方法,实现了从“单一工具”到“通用框架”的跃升,为多组学整合分析树立了新标准。相关成果以《DECODE:用于多组学数据细胞类型与细胞状态解析的通用去卷积框架》(DECODE:deep learning-based common deconvolution framework for various omics data)为题发表于《自然方法》(Nature Methods)。论文发表同期,期刊特别邀请澳大利亚科学院院士罗宾·加塞尔(Robin Gasser)团队在新闻和观点(News & Views)栏目发表专题评论,对该研究进行重点评述。
细胞类型组成及其动态状态是理解复杂生物体系和疾病发生发展的关键。组织样本中的组学数据通常来源于多种细胞的混合信号,因此需要通过去卷积(deconvolution)算法来推断不同细胞类型的比例和状态。近年来,随着转录组学、蛋白质组学和代谢组学等多组学技术的发展,大规模队列数据不断积累。然而,现有去卷积算法通常针对单一组学数据设计,难以在不同组学平台之间实现统一分析,这在很大程度上限制了多组学数据的综合利用。
针对这一挑战,王亚东教授团队开发了通用多组学去卷积算法框架DECODE。该方法能够同时适用于转录组学、蛋白质组学和代谢组学数据,并实现对细胞类型和细胞状态的联合解析,填补了代谢组学去卷积领域长期缺乏通用算法框架的空白。
研究团队构建了由多个核心模块组成的统一框架。首先,通过模拟数据生成模块构建训练数据;随后利用迁移对抗学习(transfer adversarial training)对不同组学数据进行特征对齐,从而有效消除跨平台、跨疾病状态和跨数据集之间的批次效应;进一步结合对比学习与自注意力机制对组织样本中的噪声信号进行校正和去除,实现对真实细胞特征的重建。上述模块协同作用,使DECODE在复杂数据环境下仍能稳定恢复细胞类型和细胞状态信息。
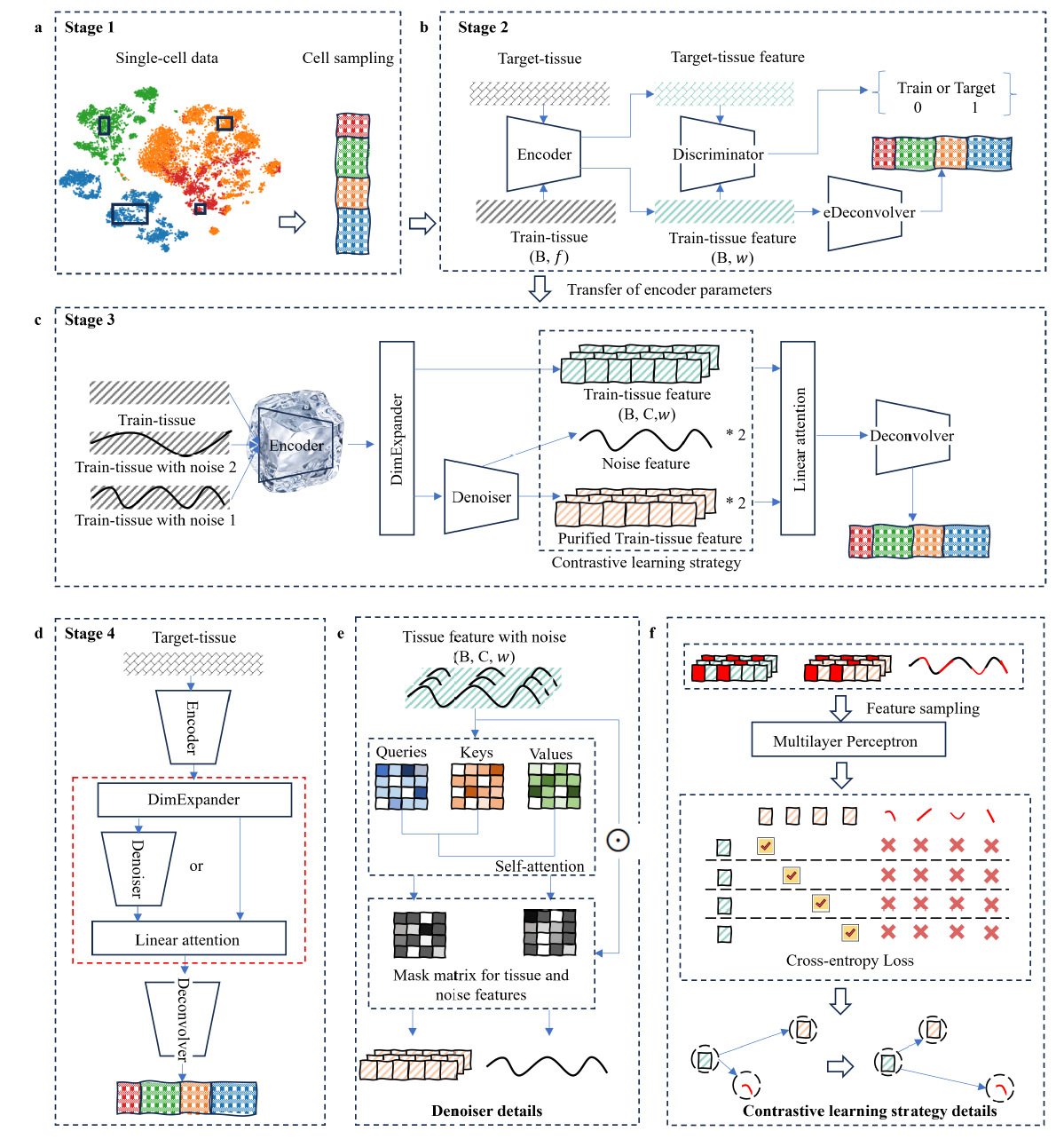
DECODE算法流程图
系统评估结果表明,DECODE在不同的组学类型、疾病状态、数据集以及测量平台下均显著优于当前主流算法,并在参考单细胞数据不完整的情况下依然能够准确解析细胞组成,表现出极高的泛化能力和鲁棒性。
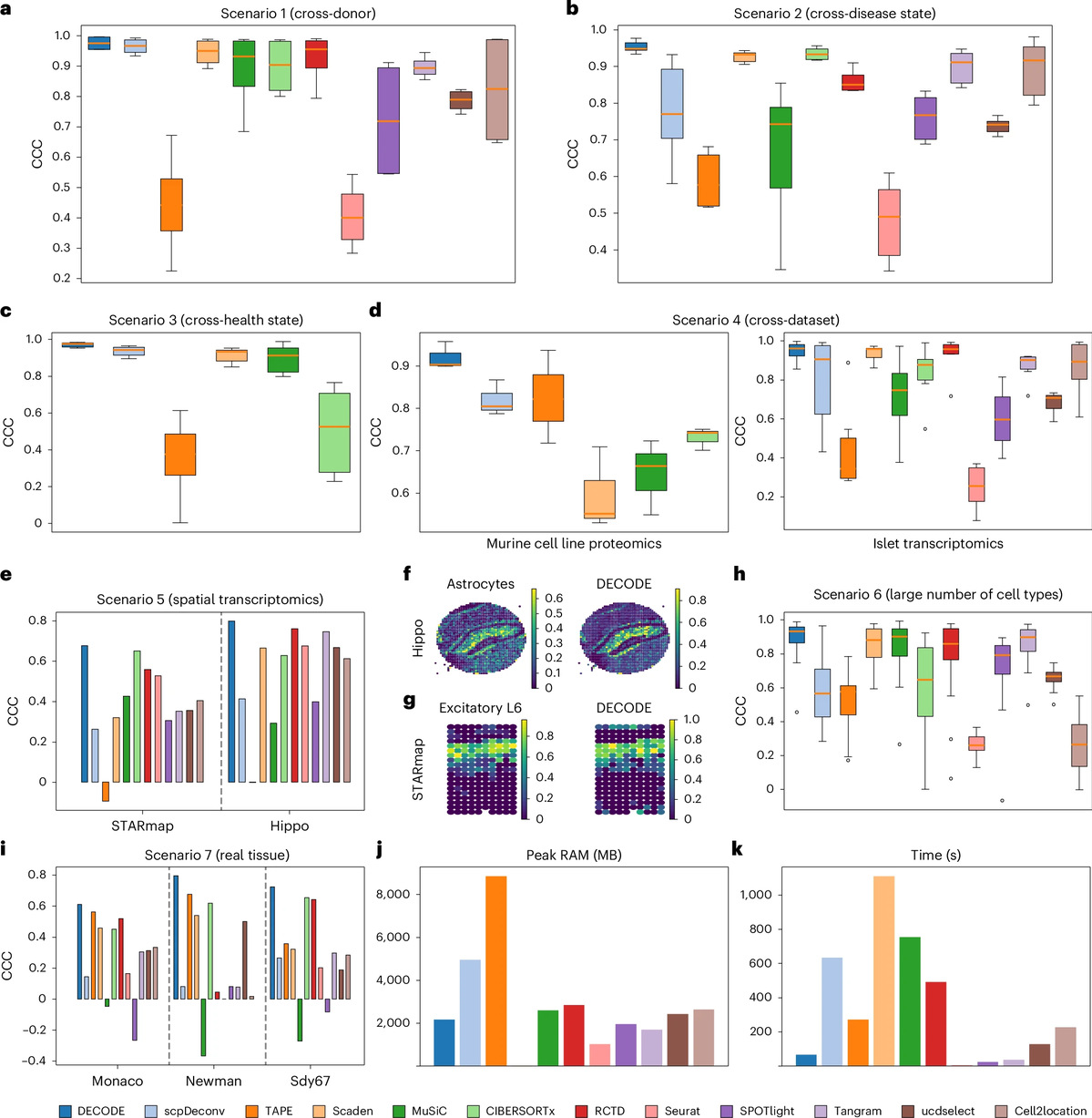
DECODE在不同组学类型、疾病状态、数据集以及测量平台下的实验结果
在乳腺癌多组学队列数据分析中,DECODE揭示了肿瘤进展过程中免疫细胞组成的重要变化。例如,在非转移性癌前病变中,T细胞比例显著升高,而B细胞比例明显降低,这一发现与既往研究中“T细胞浸润与良好预后密切相关”的结论一致。进一步的亚群分析表明,CD4⁺T细胞、CD8⁺T细胞以及增殖型T细胞在早期病变阶段均呈增加趋势,而晚期肿瘤中B细胞的增加主要来源于naive B细胞,这可能反映肿瘤免疫功能受损与转移潜能增强。
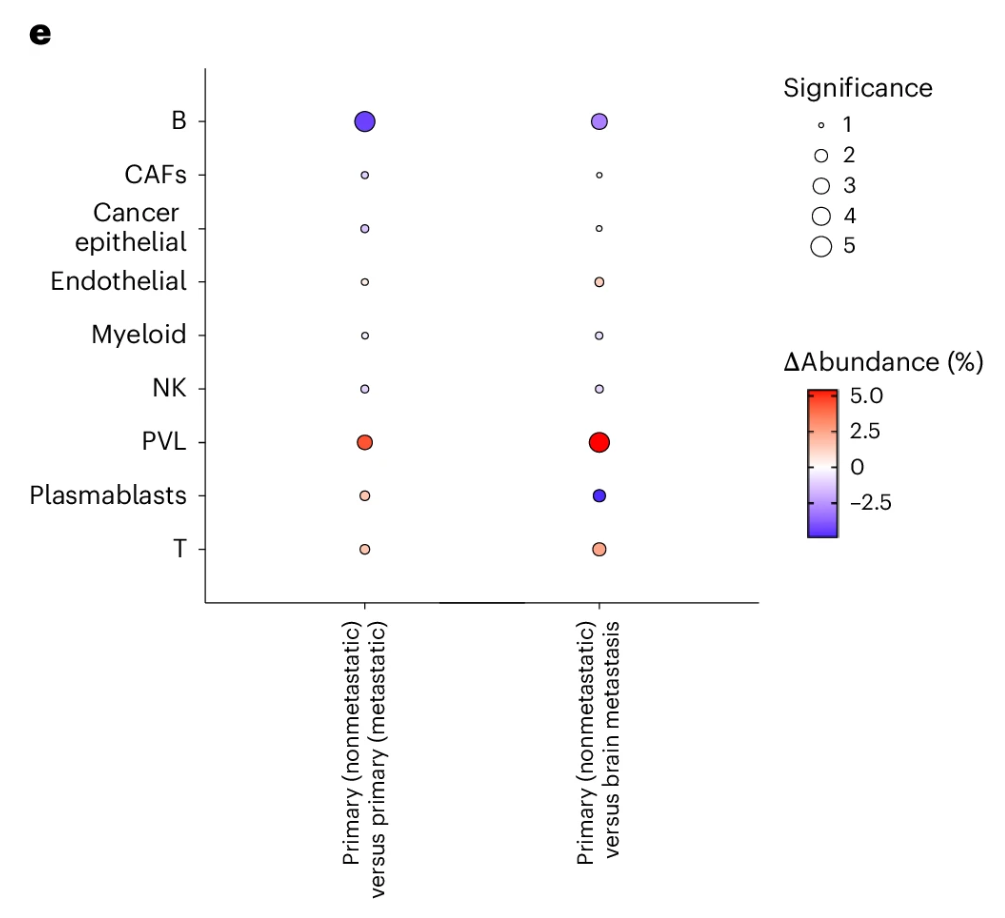
DECODE方法应用于乳腺癌数据队列的结果
该研究提出了一种通用、多组学、跨平台的细胞去卷积算法框架,不仅填补了代谢组学去卷积方法的技术空白,也为充分挖掘海量多组学队列数据提供了重要工具。DECODE有望成为连接基础研究与临床转化的重要计算平台,为疾病机制研究和精准医学提供新的技术路径。
哈工大为论文第一署名单位。王亚东教授为论文通讯作者,生命科学与医学学部赵天意教授,计算学部博士生刘任杰、孙羽志为论文第一作者。该研究获得国家自然科学基金等项目资助。
论文链接:
https://www.nature.com/articles/s41592-026-03007-y